


In pharmaceutical development, a stability study is never a mere paperwork exercise. It is the formal evidence package that links product quality to time, storage conditions, packaging, handling, and labeled use. When designed well, it supports shelf life, storage statements, expiry dating, comparability assessments, analytical method suitability, and lifecycle decisions. When designed poorly, it often leads to repeated work, fragile expiry justifications, restrictive labeling, and difficult regulatory questions during review.
For that reason, stability study design sits at the center of sound Chemistry, Manufacturing, and Controls (CMC) strategy. Under 21 CFR 211.166, the U.S. FDA expects a written program with justified test intervals, defined storage conditions, meaningful analytical methods, testing in the proposed container closure system, and, where labeling requires it, testing after reconstitution. The International Council for Harmonisation (ICH) Q1A(R2), Stability Testing of New Drug Substances and Products, remains the foundational framework for formal stability studies, while ICH Q1E, Evaluation of Stability Data, guides data evaluation for shelf life and extrapolation. FDA also published the draft guidance Q1 Stability Testing of Drug Substances and Drug Products in June 2025 as a consolidated revision of the Q1A(R2), Q1B, Q1C, Q1D, Q1E, and Q5C stability guidance.
Across the stability study designs pharma teams build, the strongest programs are designed to answer the right product question with precision, grounded in the actual dosage form, proposed market presentation, intended supply chain, likely handling conditions, and realities of the development stage.
What a Stability Study Should Actually Demonstrate
A well-designed stability program should demonstrate that a drug substance or drug product remains within acceptance criteria for identity, assay, purity, potency where relevant, physical attributes, and microbiological quality where relevant, throughout the claimed period. It should also support storage instructions and clarify what happens when the product is diluted, reconstituted, opened, thawed, shipped, or briefly exposed to conditions outside the labeled range.
In practical terms, a useful stability data definition is not simply a spreadsheet full of pull points. It forms an integrated package of evidence that includes:
- Representative batches
- The actual marketed presentation
- Stability-indicating analytical methods
- Justified storage conditions
- Meaningful pull points
- Acceptance criteria tied to specifications
- Statistical evaluation where required
- A clear conclusion regarding shelf life, retest period, or in-use period
If any one of those elements is weak, the final interpretation becomes difficult to defend.
Navigating the Regulatory Baseline
At a practical level, the regulatory question is straightforward: what should a filing-quality stability package contain? The answer is broadly consistent across the main regulatory sources:
-
21 CFR 211.166: Establishes the legal requirement for a written stability testing program to ensure drug product characteristics are maintained.
-
ICH Q1A(R2): Sets the general framework for long-term, intermediate, and accelerated studies, including storage conditions and testing frequencies.
-
ICH Q1E: Explains how data are evaluated, pooled, trended, and, where justified, extrapolated to propose a retest period or shelf life.
-
ICH Q1B: Addresses photostability testing to determine if light exposure results in unacceptable change.
-
ICH Q1D: Provides guidance on bracketing and matrixing designs to reduce the amount of testing required for multiple strengths or package sizes.
-
FDA Draft Guidance: Q1(R2) Stability Testing: The more recent FDA draft guideline which aims to bring these concepts into a more consolidated, modern structure.
-
USP <1150> Pharmaceutical Stability: Adds compendial language and practical expectations regarding storage terms, categories, and interpretation (Note: USP chapters are often behind a paywall; this link provides an overview of USP’s stability approach).
These documents form the main regulatory basis for stability study design in day-to-day practice. From the compendial side, USP stability guidelines help frame storage terminology and storage expectations alongside FDA requirements.
8 of the Most Common Pitfalls

Pitfall 1: Starting the Stability Program Too Late
Teams often wait until a filing milestone is close, then try to build a study that can answer everything at once. That approach almost always creates blind spots.
A stability program should start early enough to inform development, not merely document it. Early studies may be exploratory, but they should still clarify formulation sensitivity, degradation pathways, packaging risks, and analytical suitability. By late clinical development, the program should already support formal long-term and accelerated studies, commitment batches, and any product-specific special studies.
A better approach is to build stability planning into phase-appropriate CMC strategy. Treat early work as decision-making data and later work as registration-quality evidence. Do not assume one late study will repair months of missing context.
Pitfall 2: Using Batches That Do Not Represent the Commercial Story
Formal studies are expected to use representative primary batches, traditionally at least three for registration support. If the formulation, process, fill volume, sterilization approach, or closure system changes materially after the study starts, the original data may no longer support the story you need to tell.
A better approach is to lock the study around the version of the product you intend to file. If changes are unavoidable, document comparability logic early and decide whether bridging data, new stability batches, or a revised shelf-life proposal will be needed.
Pitfall 3: Testing the Wrong Presentation
Sponsors test material in laboratory containers, development bottles, or provisional packaging and then expect the data to support the final marketed configuration. FDA stability requirements are very clear that the product should be tested in the same container closure system proposed for marketing.
This matters because the container is part of the stability story. Headspace, stopper composition, light protection, oxygen permeability, moisture barrier properties, overwrap, orientation, and fill volume all affect results. The mistake is even more serious for sterile products, suspensions, lyophilized products, semipermeable packaging, and presentations with more than one fill size.
A better approach is to match the study samples to the intended market configuration as early as possible. If multiple strengths or container sizes are involved, justify whether full coverage is needed or whether bracketing under ICH Q1D is scientifically appropriate.
Pitfall 4: Treating the Method Panel as a Routine Release Panel
Stability studies depend on methods that can detect meaningful change over time, not just confirm that a batch met release criteria on day one.
This is where teams often underestimate forced degradation, impurity characterization, assay specificity, and physical characterization. A method that is adequate for lot release may still be inadequate for shelf-life evaluation. That is especially true for complex formulations, peptides, biologics, emulsions, suspensions, and products vulnerable to light, oxidation, or adsorption.
A better approach is to confirm that the analytical package can separate degradation from normal product signal. Use the stability study to monitor the attributes most likely to move, not just the tests that are easiest to run.
Pitfall 5: Weak Pull Point Design
A pull schedule should do more than satisfy a template. It should answer how the product changes over time and when the most informative shifts are likely to appear. Yet many protocols use inherited pull points without asking whether they match product risk, dosage form, storage state, or intended shelf life.
Poor pull design creates two opposite problems. Teams either oversample and add cost without gaining insight, or they undersample, miss an early shift, and end up with data that cannot support a clear trend.
A better approach is to choose pull points that reflect product risk and decision needs. Make sure long-term, accelerated, and intermediate conditions can be interpreted together. If a study is meant to support a meaningful shelf-life claim, the schedule should show how that claim will be defended.
Pitfall 6: Ignoring Special Conditions Until Review Questions Arrive
Many stability failures are not failures of the main long-term study. They are failures of omission: the team ran the standard chambers but did not evaluate how the product behaves in real use.
This is where product-specific studies become essential:
- freeze-thaw assessment for frozen or chilled materials
- in-use studies for multi-dose, prepared, diluted, or device-associated products
- photostability for light-sensitive presentations
- reconstitution or dilution hold time
- shipping and handling excursions
- short-term room exposure during pharmacy or clinic use
This is also where teams begin asking practical questions about the FDA room temperature definition and any defensible temperature excursion definition for shipping, pharmacy handling, or short-term clinic exposure.
A better approach is to map the full product journey from manufacture to final administration. Then ask where the product is most likely to experience stress outside the formal chamber. Design those studies before someone else asks for them. Check with pharma consultants for a better understanding.
Pitfall 7: Poor Freeze-Thaw Logic
For teams working through FDA expectations for freeze-thaw stability, the issue is simple but high-risk: can the product tolerate foreseeable handling events, or does it remain stable only under controlled, uninterrupted storage?
Freeze-thaw events matter for frozen drug substance, biologic intermediates, reference standards, and any product exposed to cold chain disruption. They can affect potency, aggregation, particle formation, viscosity, container stress, and microbial control strategy. Yet many teams run one minimal cycle without defining the true worst-case scenario.
A better approach is to set the number of cycles, hold times, thaw conditions, and test panel based on process reality. Study the material in the form and container that will experience the event. A generic one cycle exercise is rarely persuasive if the real supply chain allows repeated handling.
Pitfall 8: Superficial In-Use Studies
When teams ask what the FDA expects from in-use stability studies in practice, the focus usually falls on multi-dose vials, reconstituted products, diluted infusions, and products that remain in contact with a delivery device during administration.
The problem is usually not that teams forget these studies entirely. It is that they design them as brief laboratory demonstrations rather than as realistic simulations of labeled use.
A credible in-use study should reflect how the product is prepared, how long it remains exposed, what materials contact it, how often it is accessed where relevant, and the justified worst-case handling conditions. If the label will claim a hold time after dilution or a period after first puncture, the study should directly support that statement.
A better approach is to build the in-use protocol around the instructions for use, then challenge the design with a justified worst case. Do not assume that unopened shelf-life data automatically supports opened, diluted, or device connected use.
Pitfall 9: Misusing Bracketing and Matrixing
Bracketing and matrixing can reduce study burden, but only when the product family truly supports them. Teams sometimes apply reduced designs for convenience rather than because the formulations, strengths, fills, or container systems are suitably related.
That is risky because reduced designs trade data volume for assumptions. If variability is high, formulations differ meaningfully, or the closure system contributes to stability, those assumptions may not hold. The result can be a thinner data package and a shorter or less defensible shelf life.
A better approach is to use reduced designs only after checking formulation similarity, packaging similarity, risk, and prior knowledge. If the product is heterogeneous or the data is noisy, full testing is often the cheaper decision in the long run.
Pitfall 10: Weak Statistical Interpretation
Stability does not end when the samples are tested. The final question is whether the data justify the retest period or shelf life being claimed.
ICH Q1E exists for this reason. It addresses how stability data should be trended, when batch poolability should be assessed, when extrapolation may be considered, and how full versus reduced designs affect interpretation. Without that discipline, sponsors may overstate shelf life, ignore variability, or miss that one batch is behaving differently from the rest.
A better approach is to plan the evaluation method before the study is fully mature. Decide which attributes will be trended, what counts as significant change, whether pooling is justified, and how the final shelf-life proposal will be defended.
How Stability Study Design Should Evolve by Phase
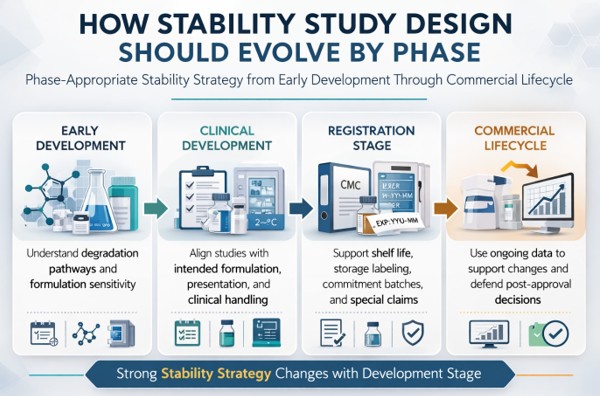
| Development Stage | What the Stability Program Should Do |
| Early development | Focus on degradation pathways, formulation sensitivity, packaging screening, and method suitability. This is the stage at which the product’s main vulnerabilities become clear. |
| Clinical development | Begin aligning studies with intended formulation, presentation, and route to filing. Shelf-life proposals become more visible, and special studies should be tied to actual clinical handling. |
| Registration stage | The program should now support expiry, storage labeling, commitment batches, and any product-specific special claims such as dilution hold time or in-use period. At this point, gaps become expensive. |
| Commercial lifecycle | Use ongoing data to confirm the labeled claim, support changes, and defend quality decisions after scale-up, site changes, or packaging modifications. |
Why Templates Are Not Enough
Templates can provide a useful starting structure, but they cannot determine the scientific content of a sound stability program. They do not decide which batches are representative, which risk points matter most, which analytical methods are truly stability-indicating, or how shelf life should be justified. A template may save formatting time, but it cannot replace technical judgment.
Our Final Thoughts on Stability Study Design
Most failures in stability study design arise from late planning, weak alignment, or omission of real-use conditions. Strong stability work is phase-appropriate, product-specific, and tied to the real product, supply chain, and label so that shelf-life claims remain defensible.

Alex has 20+ years of experience in the CMC space, specializing in CDMO/CRO management, analytical development, technology transfer, quality and regulatory compliance for various drug modalities across multiple product stages.
Reach out to Alex on LinkedIn.


