


Developing a therapeutic from lab to commercial launch requires much more than scientific innovation. it demands coordination between two pillars of the pharmaceutical business: clinical and non-clinical development. The non-clinical process demands sophisticated pharma engineering solutions that bridge laboratory promise into commercial reality.
Pharmaceutical engineering firms provide the critical infrastructure, process expertise, and validation strategies that enable biotechnology and pharmaceutical companies to navigate the journey from preclinical development through regulatory approval and commercial launch.
The following comprehensive guide explores the full landscape of pharma engineering services within the non-clinical pillar of drug development. Although, before we get there it is imperative to outline the two primary pillars of drug development.
The Pharmaceutical Development Landscape: A Two-Pillar Framework
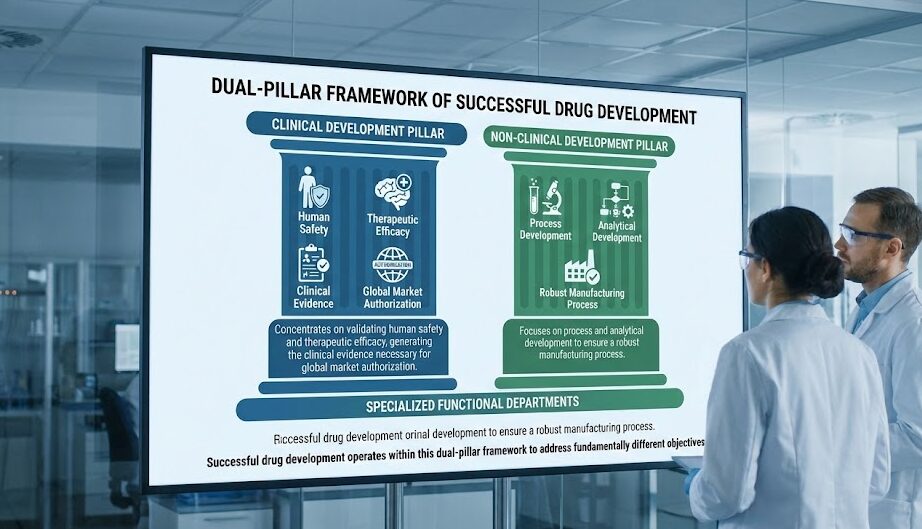
Successful drug development operates within a dual-pillar framework that addresses fundamentally different objectives through specialized functional departments. The Clinical Development Pillar concentrates on validating human safety and therapeutic efficacy, generating the clinical evidence necessary for global market authorization.
The Non-Clinical Development Pillar focuses on process and analytical development to ensure a robust manufacturing process.
This guide will focus on pharma engineering consultants that operate within the non-clinical development pillar that design the process, systems, infrastructure, and validation strategies that ensure drug manufacturability, data integrity, regulatory compliance, and operational excellence. Understanding how engineering pharma solutions integrate within this organizational framework ensures a strategic roadmap for clinical supply, development, validation and timely regulatory filings.
Clinical Development Pillar: Patient Safety and Efficacy
The clinical development pillar consists of functional departments that collectively generate human safety and efficacy evidence supporting marketing authorization applications. Clinical Development and Medical Affairs design Phase I through IV clinical programs, evaluate clinical outcomes, and author or contribute to clinical reports and research publications.
Clinical Operations execute site selection, manage the trial site, enroll patients, ensure protocol compliance, and conduct performance monitoring. Medical Safety and Pharmacovigilance monitor adverse events and safety signals throughout clinical trials and post-marketing surveillance.
Biostatistics provides the mathematical foundation for trial design and outcome analysis. Medical Writing and Regulatory Affairs transform clinical data into regulatory submissions following structured formats, utilizing content management systems engineered for CTD assembly automation. Clinical Supply Chain and Logistics manages clinical supply of investigational products under conditions preserving product integrity.
Non-Clinical Development Pillar: Safety and Drug Product Quality
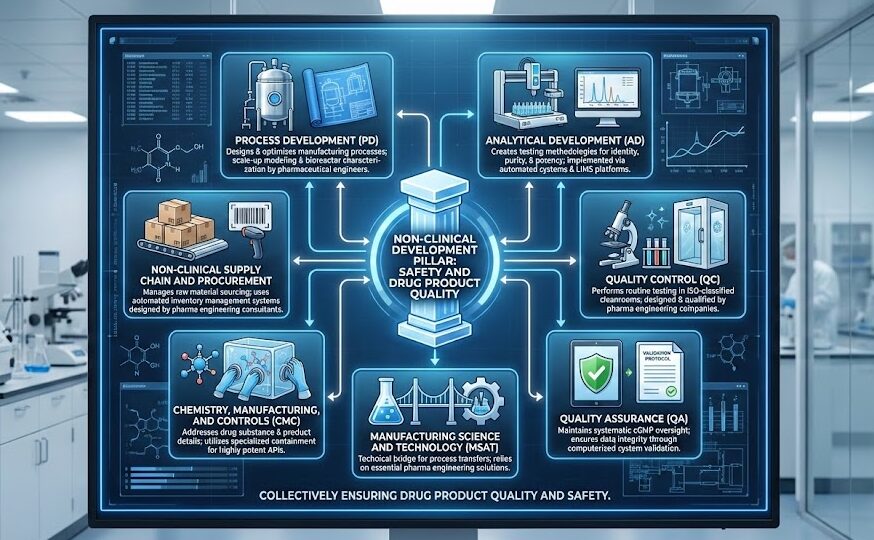
The non-clinical pillar consists of functional departments that collectively manufacture drug product and ensure product quality and safety. Process Development (PD) designs and optimizes manufacturing processes, relying on pharmaceutical engineers for scale-up modeling and bioreactor characterization.
Analytical Development (AD) creates testing methodologies for identity, purity, and potency determinations, implemented through automated liquid handling systems and integrated LIMS platforms. Quality Control (QC) performs routine testing in ISO-classified cleanrooms designed and qualified by pharma engineering companies. Quality Assurance (QA) maintains systematic cGMP oversight through computerized system validation protocols ensuring data integrity. Manufacturing Science and Technology (MSAT) serves as the technical bridge supporting process transfers, an area where pharma engineering solutions prove essential. Chemistry, Manufacturing, and Controls (CMC) addresses the drug substance and product manufacturing details, often requiring specialized containment systems for highly potent APIs.
Finally, Non-Clinical Supply Chain and Procurement manages raw material sourcing through automated inventory management systems designed by pharmaceutical engineering consultants.
The most critical pharma engineering solutions span four interconnected domains: technology transfer, infrastructure qualification, validation execution, and risk management. Each requires deep regulatory knowledge and cross-functional coordination.
Core Pharma Engineering Services: Technical Operations Excellence
Technology Transfer & Scale-Up Engineering
Technology transfer represents one of the most critical and risk-prone steps in pharmaceutical development. Pharma engineering consultants lead tech transfer projects by generating detailed process descriptions followed by comprehensive facility fit assessments that evaluate whether receiving sites possess the equipment capabilities, utility capacities, and environmental controls necessary to execute transferred processes. These assessments identify gaps between development-scale operations and commercial manufacturing requirements, enabling proactive mitigation strategies.
Scale-up engineering predicts how laboratory-optimized processes will perform in pilot-scale and commercial-scale vessels. Engineering pharma specialists develop detailed tech transfer packages that include equipment specifications, operating parameter ranges with scientifically justified ranges, and risk assessments identifying critical process parameters that require enhanced monitoring and control during manufacturing campaigns.
GMP Infrastructure & Equipment Engineering
Pharmaceutical manufacturing demands highly controlled environments and specialized equipment designed to meet stringent regulatory standards. Pharma engineering companies provide expertise in designing ISO-classified cleanroom suites with validated HVAC systems, pressure cascade controls, and environmental monitoring programs.
Equipment specifications must align with User Requirements Specifications (URS) that translate process needs into engineering deliverables. The Commissioning, Qualification, and Validation (CQV) master plan serves as the strategic framework governing how facilities, systems, and equipment are designed, installed, tested, and operated according to regulatory requirements and industry standards. This master plan establishes the validation philosophy, defines equipment classification systems determining qualification rigor, outlines documentation standards, and creates the validation schedule coordinating equipment qualification with process validation timelines.
Pharmaceutical engineering firms develop these CQV master plans to ensure regulatory inspectors can quickly understand the facility’s validation approach and verify that all GMP-critical systems have received appropriate qualification attention.
Validation Strategy & Execution
Equipment qualification follows the traditional IQ/OQ/PQ progression, with Installation Qualification verifying correct installation per manufacturer specifications and design drawings, Operational Qualification confirming equipment operates within specified parameters across the operating range, and Performance Qualification demonstrating consistent performance under actual production conditions.
Process validation, governed by FDA’s 2011 Process Validation Guidance and ICH Q11, employs a lifecycle approach encompassing process design (Stage 1), process qualification including Process Performance Qualification or PPQ (Stage 2), and continued process verification (Stage 3). The PPQ protocol represents the culmination of development activities, demonstrating that the commercial manufacturing process consistently produces material meeting predetermined quality attributes.
Analytical method validation ensures testing procedures accurately measure critical quality attributes, following ICH Q2(R2) requirements for specificity, accuracy, precision, linearity, range, and robustness. Pharma consultants and more specifically pharma engineering consultants coordinate these interconnected validation activities to prevent timeline conflicts and ensure validation data packages support regulatory submissions.
Risk Assessment & Specialized Operations
Manufacturing risk assessment frameworks, aligned with ICH Q9 Quality Risk Management principles, systematically identify potential failure modes affecting product quality, patient safety, or process capability.
Engineering pharma specialists facilitate Failure Mode and Effects Analysis (FMEA) workshops that score risks based on severity, occurrence probability, and detectability, generating prioritized mitigation strategies. Quality risk assessment extends beyond manufacturing to encompass analytical methods, facility systems, and supply chain vulnerabilities.
Specialized operations present unique validation challenges requiring deep pharmaceutical engineering expertise. Fill-finish validation for sterile operations demands container-closure integrity testing, media fill simulation studies demonstrating aseptic process capability, and environmental monitoring programs confirming Grade A/B classification maintenance throughout filling operations.
Cold chain qualification and validation ensures temperature-sensitive biologics maintain stability during storage and distribution, involving thermal mapping studies, shipping container qualification across seasonal temperature extremes, and temperature excursion protocols defining acceptable deviation limits. Environmental monitoring programs track viable and non-viable particulates, ensuring manufacturing environments continuously meet classification requirements between formal re-certification cycles.
Pharma Engineering Solutions Across Clinical Development Operations
Regulatory agencies scrutinize facility descriptions, equipment validation summaries, and process validation reports with increasing rigor as therapeutic complexity grows. FDA CMC reviewers expect Process Performance Qualification data demonstrating commercial process capability, comprehensive CQV master plans evidencing systematic validation approaches, and risk assessments justifying control strategies for critical quality attributes.
Inadequate pharma engineering planning generates costly downstream consequences: tech transfer failures requiring process re-optimization delay clinical supply; validation protocol deficiencies trigger regulatory Information Requests extending review timelines; facility fit gaps necessitate capital-intensive equipment retrofits.
Pharmaceutical engineering consultants accelerate market authorization by ensuring validation strategies align with regulatory expectations from project inception, facility fit assessments prevent infrastructure surprises during scale-up, and equipment specifications incorporate lessons learned from comparable products. This proactive engineering approach compresses development timelines, reduces regulatory friction, and positions companies for first-cycle approval success.
Partner With Pharma Engineering Experts
DES Pharma Consulting brings deep CMC expertise across both non-clinical technical operations and clinical development infrastructure.
Whether you’re facing facility fit challenges during tech transfer, need CQV master plan development for your first PPQ campaign, or require cold chain validation for gene therapy distribution, our pharmaceutical engineering solutions are tailored to your development stage and therapeutic modality. Contact us to discuss how expert pharma engineering accelerates your path to regulatory approval.

Matt specializes in Process Development (PD) and Chemistry, Manufacturing, and Controls (CMC) strategy, helping life sciences companies navigate early-stage development, lab innovation, and technical regulatory hurdles.
Reach out to Matt on LinkedIn.


