


Analytical Development vs Process Development: Integrated CMC Strategy Across Drug Development Phases
In pharmaceutical and biotechnology drug development, analytical development vs process development represents one of the most critical functional partnerships within CMC (Chemistry, Manufacturing, and Controls) operations. While both functions operate under the CMC umbrella, they serve distinct, yet reciprocally dependent roles across the development lifecycle.
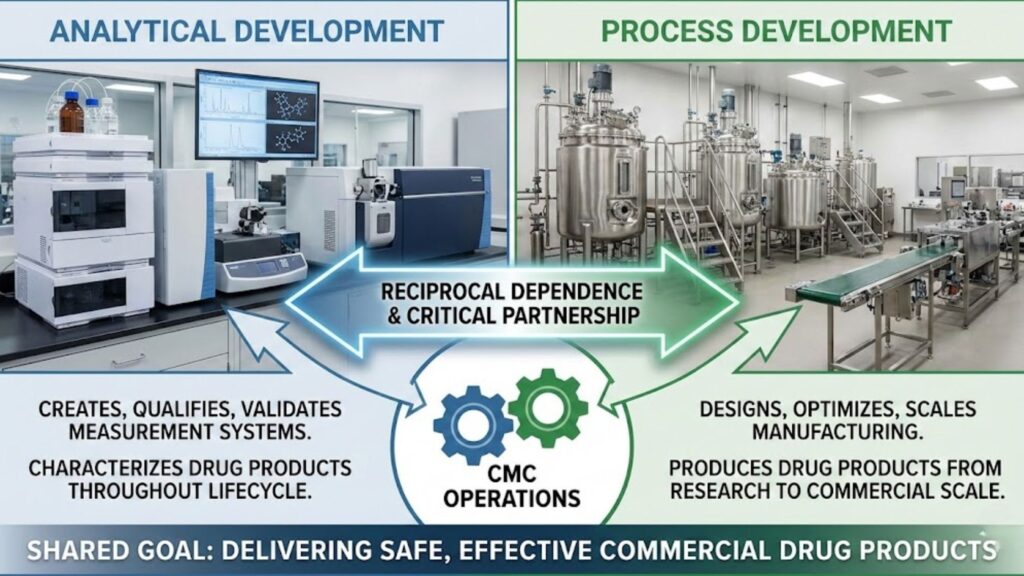
Analytical development creates, qualifies, and validates the measurement systems that characterize drug products throughout their lifecycle. Whether driven by internal teams or guided by expert pharma consulting, process development operations, by contrast, design, optimize, and scale the manufacturing processes that produce those drug products from research-scale to commercial production.
This guide clarifies the responsibilities, deliverables, and integration points between analytical development and process development methodology across three distinct development phases: Discovery to IND, IND Clearance to End-of-Phase 2, and Pivotal Trials to Commercial Launch. Understanding this integration is essential for pharmaceutical technical operations leaders, CMC directors, and process engineer R&D roles tasked with delivering regulatory-ready programs for biotechnology drugs.
Defining Analytical Development vs Process Development
Pharmaceutical analytical development encompasses the scientific discipline of creating and maintaining measurement systems that quantify quality attributes throughout drug development.
What Exactly Is Analytical Development?
Understanding what is analytical development begins with recognizing its core responsibilities:
- Analytical method development: Creating assays for strength, titer, potency, purity, identity, aggregates, and product-related impurities specific to the drug modality (monoclonal antibodies, antibody-drug conjugates, gene therapies, cell therapies)
- Method qualification and validation: Establishing method performance per ICH Q2(R2) requirements including specificity, linearity, accuracy, precision, range, and robustness
- Specification strategy: Developing acceptance criteria based on analytical trending data and regulatory expectations
- Reference standard generation and qualification: Creating primary and working reference materials with documented traceability
- Method transfer to Quality Control: Deploying analytical methods to GMP testing laboratories with statistical acceptance criteria (USP <1220>)
- Comparability analytics: Executing analytical bridging studies when manufacturing processes change per ICH Q5E guidance
- Lifecycle of Analytical procedures: Ensuring that analytical procedures remain fit for their intended purpose through scientific risk-based approach development ICH Q14, Q2(R2), validation, implementation and ongoing performance verification
Analytical development ensures that measurement systems are fit-for-purpose, validated for their intended use, and capable of supporting regulatory submissions from IND through BLA filing.
Process Development Functions
Process development methodology encompasses manufacturing process design, optimization, and scale-up. A process engineer R&D working within process development executes:
- Cell line and expression system development: Selecting expression platforms, generating Master Cell Banks (MCB) and Working Cell Banks (WCB), establishing cell line characterization strategies
- Drug substance upstream process design: Optimizing cell culture conditions including media selection, bioreactor parameters (temperature, pH, dissolved oxygen, agitation), seeding density, and harvest strategies
- Drug substance downstream process design: Developing purification unit operations including depth filtration, chromatography, tangential flow filtration (TFF), viral clearance (inactivation and filtration)
- Drug product formulation and fill-finish: Designing formulation composition (buffer systems, stabilizers, excipients, concentration), container closure systems, sterile filtration, and filling processes
- Process characterization: Identifying Critical Quality Attributes (CQAs) and Critical Process Parameters (CPPs), establishing design spaces through Design of Experiments (DOE), determining Normal Operating Ranges (NOR) and Proven Acceptable Ranges (PAR)
- Scale-up and technology transfer: Translating laboratory-scale processes to pilot and commercial scale, executing tech transfer to Contract Manufacturing Organizations (CMOs)
Process development delivers the manufacturing blueprint that produces drug substance and drug product meeting predefined quality specifications.
The Integration Imperative
Analytical development and process development share fundamental dependencies that require coordinated execution:
Process development depends on analytical development to measure whether process optimization efforts improve or degrade product quality. Without validated analytical methods, process changes cannot be assessed for their impact on CQAs.
Analytical development depends on process development to generate material for method development, qualification, and validation. Engineering batches, GMP batches, and process characterization samples provide the matrix needed for analytical method robustness testing.
Both disciplines depend on Quality by Design (QbD) integration to connect product requirements to manufacturing controls and analytical measurement. The qbd process establishes the framework:
1. Quality Target Product Profile (QTPP): Defines target product characteristics (dosage form, route of administration, stability requirements, performance attributes)
2. Critical Quality Attributes (CQAs): Product parameters impacting safety or efficacy, identified through risk assessment
3. Critical Process Parameters (CPPs): Manufacturing inputs controlling CQAs
4. Analytical strategy: Methods measuring CQA variability within the CPP design space, incorporating Analytical Target Profile (ATP) and Method Operable Design Region (MODR) concepts per ICH Q14
A well-structured qtpp template explicitly links each CQA to the analytical method measuring it and the process parameters controlling it, creating an integrated control strategy that demonstrates process understanding to regulatory agencies. This analytical qbd approach ensures that measurement systems can detect product variability across the manufacturing design space while process development pharmaceutical teams maintain tight control over critical parameters.
Phase-Specific AD and PD Deliverables
Discovery to IND
Scope: Lead Candidate Identification → IND “Safe to Proceed” Status
Phase Objective: Transition from laboratory concept to regulated entity ready for human testing through pre-clinical research, candidate optimization, process scouting for GLP toxicology material, and establishing initial safety profile bridging research to regulated manufacturing. This phase is critical in biotechnology and drug discovery programs.
Analytical Development Deliverables:
- Testing panel development: Modality-specific analytical package covering titer, potency, purity, identity, characterization assays for CQA baseline (primary structure, potency, purity, aggregates)
- Method development for impurities: Process-related and product-related impurity assays
- Stability-indicating method development: Preliminary stress testing to demonstrate degradation sensitivity
- Specification strategy: Small-scale batch analytical trending, preliminary acceptance criteria development
- Method transfer: Testing panel deployment and Method Transfer Report
- Analytical comparability assessment: Toxicology vs Phase 1 clinical batch compatibility studies with analytical risk assessment (FMEA) and comparability study execution
- Reference material strategy: Engineering batch reference material generation, GMP reference material initial qualification
- IND Module 3.2.S/3.2.P: Analytical regulatory sections, methods descriptions, justifications, and rationale for phase-appropriate method qualification (vs full validation)
Process Development Deliverables:
- Cell line development: Expression system definition, cell line selection, Research Cell Bank (RCB) and Master Cell Bank (MCB) generation with scale-up strategy, transition from FBS-containing media
- Drug substance process design: Upstream scale-up/scale-out (T-flask → shake flasks → bioreactor), harvest strategy development; Downstream scale-up/scale-out with robust scalable unit operations, buffer system selection
- Drug product process design: Closed, sterile, scalable unit operations, preliminary formulation screening (stabilizer/excipient screening, concentration range-finding), container closure suitability assessment
- Material generation: GMP-representative small-scale batches, engineering batch manufacturing (non-GMP), GMP clinical batch execution, reference material strategy for scale changes
- CMO qualification: CMO selection, tech transfer planning, GMP readiness assessment
- IND Module 3.2.S/3.2.P: CMC regulatory sections, rationale for phase-appropriate in-process controls
IND Clearance to End-of-Phase 2
Scope: IND Clearance → End-of-Phase 2 (EOP2) Meeting
Phase Objective: Execute Phase 1 and Phase 2 clinical trials emphasizing human safety and dose-finding; GMP manufacturing of clinical trial material; scale-up from lab to pilot scale; fine-tuning formulation before pivotal trials; demonstrating clinical proof-of-concept and building scalable manufacturing processes. This represents a critical transition point in biotechnology in drug discovery where r&d process engineer teams refine both analytical and process capabilities.
Analytical Development Deliverables:
- Method qualification: Non-platform and characterization assay qualification, forced degradation studies
- Analytical Target Profile (ATP) development: Method performance requirements (LOD/LOQ, range, precision, robustness), preliminary robustness and intermediate precision assessment
- Formal method transfer: Comparative testing protocols between development and GMP labs with statistical equivalence analysis
- GMP analytics deployment: All GMP-ready methods for batch release, system suitability testing (SST), OOS investigation procedures
- Reference material hierarchy: Primary and working reference standard qualification, traceability, comparability, stability monitoring program, inventory forecasting
- Specification refinement: Analytical data trending strategy, cumulative GMP batch data analysis, specification acceptance criteria development
- Comparability management: Analytical comparability assessment for scale-up, formulation optimization, site transfer; comparability study design with statistical analysis; method variability management
- Control of Critical materials: Critical reagent and column qualification, alternate vendor assessment
- System for Management of Laboratory Data: LIMS implementation, analytical data integrity strategy
- Extended characterization: Intermediate and DS/DP freeze-thaw studies, container/closure compatibility studies, characterization panel validation, orthogonal potency assay verification
- Method lifecycle management: ICH Q14 continuous verification strategy of Analytical procedures
Process Development Deliverables:
- Cell bank generation: Working Cell Bank (WCB) generation, MCB and WCB characterization (identity, purity, stability), expression system characterization and qualification
- Upstream optimization: DOE for design space mapping, seeding density optimization, seed train optimization, media optimization, production optimization (fed-batch, perfusion), reactor parameter design space (temperature, agitation, dissolved oxygen, pH), metabolic profiling, worst-case/edge-of-failure studies
- Downstream optimization: Depth filter optimization (flocculation agent screening, flux rate, capacity), chromatography optimization (residence time, bed height, buffer optimization, dynamic binding capacity, pooling criteria), TFF optimization (crossflow flowrate, flux, shear, concentration factor, diafiltration volumes), viral clearance development (filter selection, inactivation strategies)
- Process attribute profiling: Characterized range determination, preliminary NOR determination, intermediate hold mapping
- Drug product optimization: Filling equipment selection and qualification, container closure format selection, CCS material and system selection, formulation design space (pH/buffer optimization, concentration optimization, dose-form selection), filtration and filling parameter optimization
- Quality by Design implementation: CQA identification and risk ranking, process and material parameter risk assessment, pre-Process Characterization risk assessment, scale down model qualification protocol, process robustness studies
- Tech transfer to CMO: Platform development, tech transfer planning, initiation, and completion
- Comparability strategy: Process impurity trending, product quality risk assessment, process capability trending, process and analytical comparability protocol development and execution, optimized process scale-down batches
- Regulatory support: IND amendment CMC authoring
Pivotal Trials to Commercial Launch
Scope: Phase 3 Initiation → Market Authorization & Lifecycle Management
Phase Objective: Late-stage development and transition to long-term commercial product; process validation through PPQ batches; NDA/BLA Module 3 CMC compilation; Pre-Approval Inspection (PAI) preparation; post-market lifecycle management including post-approval changes, site transfers, and COGS optimization.
Analytical Development Deliverables:
- Method validation completion: Full ICH Q2(R2) validation parameters (specificity, linearity, accuracy, precision, robustness) for all release and stability methods, orthogonal potency assay validation, characterization panel validation
- Validation documentation: Analytical Validation Master Plan, Method Validation Reports, Analytical Robustness Qualification Reports, In-Process Sample Qualification Reports, Material Release Method Validation
- Extractables/leachables studies: E/L studies for commercial container/closure system
- Analytical PPQ: Analytical PPQ Protocol, statistical analysis of PPQ analytical data, analytical portions of PPQ report, supporting justification of specifications with PPQ datasets
- Reference standards for commercial operations: Multi-site reference material strategy, Reference Material Characterization Validation, commercial reference standard requalification protocol and execution schedule, commercial reference material lifecycle management strategy
- Commercial specifications: Robust analytical trending data package, justification of specifications, analytical control strategy finalization
- Material and equipment management: Equipment requirements and performance criteria, equipment change control technical assessment, material and equipment lifecycle planning, commercial equipment tech transfer protocols
- PAI readiness: Analytical method validation package review, raw data audit, OOS investigation documentation, mock PAI analytical audit and gap remediation
- Lifecycle management: Analytical continuous improvement program, post-approval analytical change management strategy, analytical comparability protocols for post-approval changes, Annual Product Review (APR) analytical data compilation and trending, analytical method transfer and site transfer protocols
Process Development Deliverables:
- Cell bank qualification: MCB and WCB qualification and stability, WCB comparability and replacement strategy
- Process characterization completion: Drug substance upstream PC (unit operation characterization, inoculum expansion characterization, limit of in vitro age determination, MCB/WCB comparability, expression system storage validation), drug substance downstream PC (unit operation characterization, impurity spiking, aged resin characterization, resin lifetime and carryover evaluation), drug product PC (bulk formulation hold-time studies, filtration studies, lyophilization and reconstitution studies, E/L risk assessment, final formulation justification)
- Post-PC risk assessments: Drug substance and drug product post-PC risk assessments, raw material risk assessment, consumable risk assessment
- Process validation: Process Performance Qualification (PPQ) protocol development, PPQ sampling plan, commercial sampling plan, Process Validation Master Plan (PVMP) support, validation protocol support, validation execution technical support
- Control strategy development: Contamination Control Strategy (CSS) development, commercial process control strategy, Normal Operating Range (NOR) and Proven Acceptable Range (PAR) determination
- Commercial process lock: Scale down model qualification, N-determination, quality attribute determination, in-process attribute level (IPAL) determination, parameter classification, viral clearance validation, commercial process description, commercial process implementation and lock, foundation building for future process changes
Our Final Thoughts on PD vs AD
Integrated analytical development and process development pharmaceutical strategies accelerate biotechnology drugs programs from IND submission to BLA approval by preventing costly CMC delays rooted in functional misalignment. Phase-appropriate analytical and process maturity—methods qualified at IND, progressing toward validation during Phase 2, fully validated at BLA—ensures regulatory expectations are met without over-investment in early development or under-investment approaching commercial launch.
The qbd process framework connecting QTPP to CQA to CPP to analytical strategy provides the structure for demonstrating process understanding and analytical control to regulatory agencies. The analytical qbd principles including ATP and MODR development ensure methods remain fit-for-purpose throughout manufacturing scale-up and lifecycle management, supporting both pharmaceutical analytical development and process development methodology execution.
Comparability planning executed before process changes—with pre-defined analytical acceptance criteria per ICH Q5E—protects clinical data continuity and prevents regulatory delays. Understanding analytical development vs process development, and how these functions interlock across development phases, represents essential knowledge for CMC directors, VPs of Technical Operations, and process engineer r&d professionals responsible for delivering regulatory-ready programs across biotechnology in drug discovery.
Partner with DES Pharma Consulting
DES Pharma Consulting provides analytical development and process development & CMC consulting services for biologics programs from pre-clinical through BLA filing. We help pharmaceutical and biotechnology companies design integrated AD/PD roadmaps, develop comparability protocols, and provide technical oversight for CMO operations. Contact DES Pharma Consulting to discuss how we can support your analytical and process development integration needs.

Senior Consultant, Process Development & CMC

