

For AAV gene therapy programs, two analytical questions sit at the center of every CMC discussion with regulators: how much vector is in the vial, and does it work? These questions are answered through vector genome (VG) titer and potency assays — and the methods chosen, the way they’re qualified, and the way they’re validated will shape every release decision, every comparability study, and every IND, IND amendment, and BLA the program submits.
This guide walks through the analytical landscape for AAV titer and potency, the strengths and limitations of each major method, and what FDA and EMA expect for method validation as a program advances from IND-enabling work through commercial.
Why Titer and Potency are Uniquely Consequential for AAV
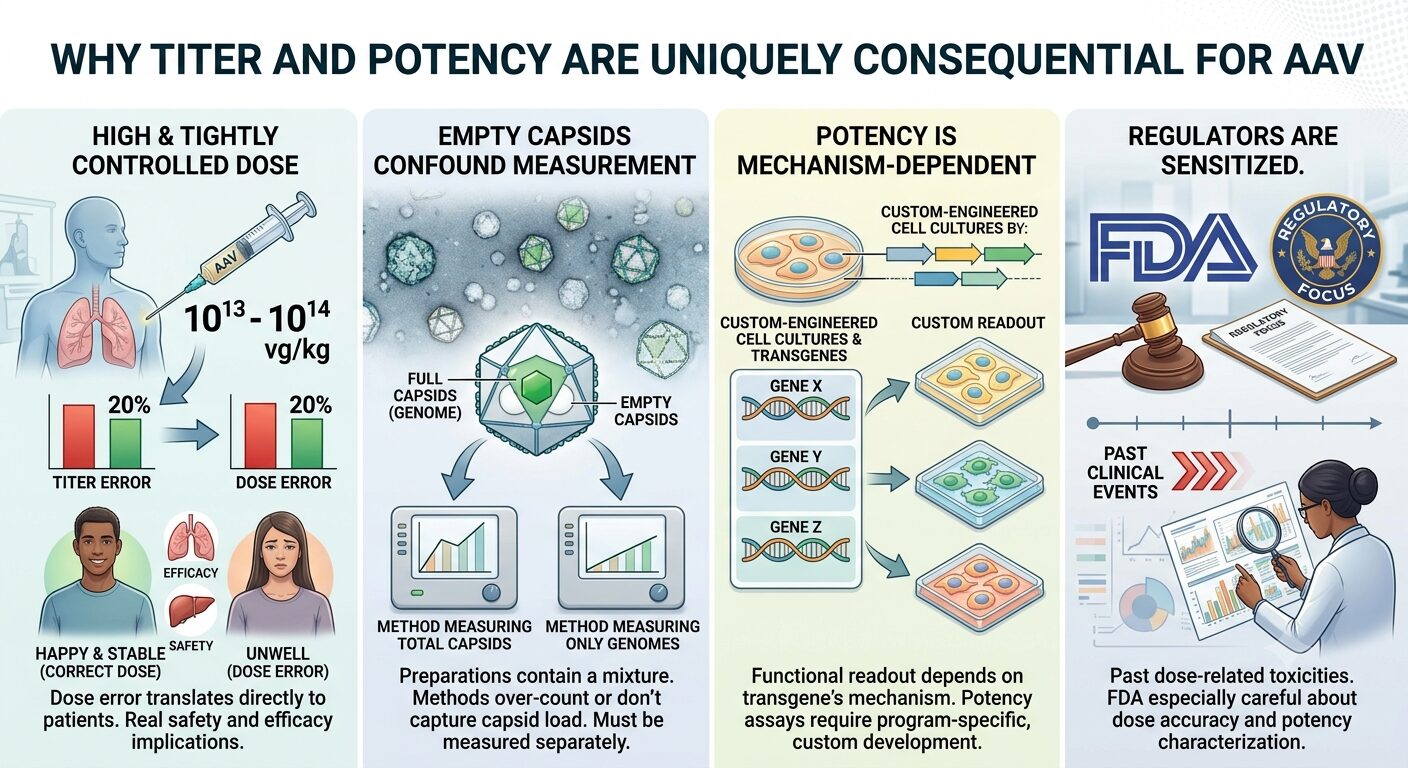
For most biologics, titer and potency are important but well-established analytical problems. For AAV, both are unusually difficult and unusually consequential.
- Dose is high and tightly controlled. AAV doses are often in the 10¹³ – 10¹⁴ vg/kg range. A 20% error in titer measurement translates directly to a 20% dose error in patients, with real safety and efficacy implications.
- Empty capsids confound measurement. AAV preparations contain a mixture of full (genome-containing) and empty capsids. Methods that measure total capsids over-count; methods that measure only genomes don’t capture capsid load. Both matter, and they must be measured separately.
- Potency is mechanism-dependent. The functional readout for an AAV depends on the transgene’s mechanism of action, which means potency assays are usually program-specific and require custom development.
- Regulators are sensitized. Past clinical events involving dose-related toxicities have made FDA especially careful about AAV dose accuracy and potency characterization.
Vector Genome Titer: the Dominant Methods
Droplet digital PCR (ddPCR)
ddPCR has emerged as the dominant method for AAV vector genome titer in most modern programs. The method partitions the sample into thousands of nanoliter droplets, performs PCR in each, and counts positive vs negative droplets to give an absolute quantification without a standard curve.
Strengths:
- Absolute quantification without reliance on a calibration curve
- Excellent precision and reproducibility
- Tolerant of PCR inhibitors common in AAV preparations
- Increasingly accepted by FDA as a primary release method
Limitations and pitfalls:
- Sensitive to assay design — primer/probe placement matters significantly
- Linearization of the genome before PCR is critical; encapsidated genome conformation affects results
- Sample treatment (DNase digestion, capsid lysis) must be carefully optimized and consistent
- Requires specialized instrumentation (Bio-Rad QX series; Stilla; others)
Quantitative PCR (qPCR)
The historical standard for AAV genome titer. qPCR remains in widespread use, particularly for legacy programs and where ddPCR is unavailable.
Strengths:
- Widely available, mature instrumentation
- Lower cost per sample than ddPCR
- Long-established methodology with extensive regulatory precedent
Limitations:
- Relies on a standard curve, which must itself be carefully prepared and bridged across lots
- More sensitive to inhibitors than ddPCR
- Higher inter-lab variability historically reported
- Increasingly being supplanted by ddPCR for primary release testing
Optical/spectroscopic methods
For total capsid quantification (distinct from vector genome titer), methods include:
- ELISA-based capsid titer using serotype-specific anti-capsid antibodies
- UV spectroscopy at A260/A280 for full vs empty determination in purified preparations
- Charge-detection mass spectrometry (CDMS) for individual particle mass measurement
- Analytical ultracentrifugation (AUC) as the longstanding orthogonal method for full:empty ratio
Method comparison summary
| Method | What it measures | Typical use | Regulatory acceptance |
|---|---|---|---|
| ddPCR | Vector genome titer (vg/mL) | Primary release method | Broadly accepted |
| qPCR | Vector genome titer (vg/mL) | Release method, increasingly secondary | Accepted, mature |
| ELISA (capsid) | Total capsid titer (cp/mL) | Capsid quantification, full:empty calculation | Accepted as orthogonal |
| AUC | Full:empty ratio, aggregation | Characterization, comparability | Widely used, increasingly expected |
| CDMS | Mass distribution at single-particle level | Characterization, emerging release use | Emerging acceptance |
| Cryo-EM | Capsid integrity, full:empty visualization | Characterization | Characterization-grade |
| SEC-MALS | Aggregation, size distribution | Purity, stability | Accepted as orthogonal |
Potency: The Harder Problem
Potency assays for AAV must demonstrate biological function in a way that’s relevant to the mechanism of action and stability-indicating. FDA’s expectation, articulated in guidance on potency tests for cellular and gene therapy products, is that potency be measured by a method that reflects the mechanism by which the product produces a therapeutic effect.
For AAV, this typically means a cell-based functional assay rather than a surrogate measurement. The major categories:
Transduction-based potency assays
These measure the ability of the vector to transduce cells and express the transgene. A susceptible cell line (often HEK293, HeLa, or a cell type relevant to the target tissue) is exposed to a dilution series of vector, and transgene expression is quantified by:
- Reporter readout (luciferase, GFP) when the transgene includes a reporter
- RT-qPCR or ddPCR for transgene mRNA
- Protein expression by ELISA or Western blot
- For secreted protein transgenes, ELISA on conditioned media
Function-based potency assays
These go beyond expression to demonstrate that the expressed protein performs its biological function. Examples:
- Enzymatic activity assays for therapeutic enzymes (e.g., AAV expressing iduronidase measured by enzymatic substrate cleavage)
- Bioactivity assays measuring the downstream cellular consequence of transgene expression
- For gene editing payloads (Cas9, base editors), measurement of editing activity at the target locus
FDA increasingly prefers function-based assays where feasible, because they more directly reflect therapeutic mechanism. A transduction assay that shows GFP expression doesn’t prove the therapeutic transgene will function correctly in patients.
Matrix approach: when one assay isn’t enough
For complex products, regulators increasingly accept and even prefer a “potency matrix” — multiple complementary assays that together characterize the functional profile. For an AAV with a complex mechanism, the matrix might include:
- A genome titer assay (to confirm dose delivery)
- A transduction assay (to confirm cell entry and basic expression)
- A function assay (to confirm therapeutic activity)
- A stability-indicating assay (often the same as one of the above, but specifically demonstrated to track potency loss over time)
Establishing a robust analytical control strategy is core to analytical method development and validation, and for AAV programs this work disproportionately determines program risk.
Method Validation: What Regulators Expect
Method validation for AAV titer and potency follows the general principles of ICH Q2(R2), with AAV-specific considerations layered on. The validation parameters expected for a primary release method:
| Parameter | Titer (e.g., ddPCR) | Potency (cell-based) |
|---|---|---|
| Specificity | Required — specific for the transgene/regulatory sequence | Required — specific for intended biological activity |
| Accuracy | Required — recovery against reference standard | Difficult; demonstrated by parallelism and relative potency |
| Precision (repeatability, intermediate, reproducibility) | Required at all levels | Required; cell-based assays typically show higher variability |
| Linearity / Range | Required across the working range | Required across the working range; relative potency methodology |
| LOD / LOQ | Required if relevant to specification | Required if relevant to specification |
| Robustness | Required — sample prep, instrument, reagent lot effects | Required — cell passage, reagent lot, operator effects |
| System suitability | Required — defined criteria each run | Required — defined criteria each run |
Stage-appropriate validation: matching effort to phase
Methods don’t need to be fully validated at first-in-human. The expectation scales with development stage:
- Pre-IND / IND-enabling: Methods qualified for intended use, with documented performance and a validation plan
- IND / Phase 1: Qualified methods used for release; validation may be in progress, with key parameters demonstrated
- Phase 2: Validation substantially complete for release methods; characterization methods may remain qualified
- Phase 3 / PPQ: Full validation for all release and stability-indicating methods, with method transfer to commercial QC labs complete
- BLA submission: Validation reports for all release methods; performance history demonstrated through clinical lots
Pushing validation effort too late is a common late-stage CMC pitfall. By Phase 3, validation needs to be complete; if methods are still being optimized when PPQ runs are executed, the comparability question becomes much harder to resolve cleanly.
Reference standard strategy
For AAV, the reference standard is the linchpin of every quantitative measurement. A robust reference standard strategy involves:
- Primary reference standard — a well-characterized lot of the actual product (or a representative version), stored under defined conditions, with detailed characterization
- Working reference standards — qualified against the primary, used for routine analysis to preserve the primary for critical bridging studies
- Bridging plan — how successor reference standards are qualified against current ones, with overlap testing
- Stability program — periodic re-qualification of reference standards against historical performance
- External standards where available — for AAV2 and AAV8, the AAV Reference Standard Material program (ATCC) provides community standards that can be used as orthogonal references in method development
Sponsors that skimp on reference standard characterization create problems that compound across the program. Every titer measurement you make is ultimately a measurement against your reference standard — if that standard’s value is uncertain, all your measurements inherit that uncertainty.
6 Common pitfalls in AAV analytical programs
- Underinvested potency assays. Teams develop a transduction reporter assay and consider potency “done.” Regulators frequently push for mechanism-relevant function assays in later phases.
- No orthogonal methods for capsid characterization. Reliance on a single full:empty method (e.g., AUC alone) without an orthogonal confirmation (CDMS, cryo-EM) leaves uncertainty unaddressed.
- Method drift across the program. Subtle changes to sample preparation, reagent lots, or instrumentation accumulate, creating apparent product variability that’s actually method variability.
- Inadequate cell bank management for cell-based assays. Cell passage number, media changes, and cell line authentication all affect potency assay performance.
- Reference standard scarcity. Running through primary reference standard inventory before a successor is qualified.
- Validation deferred until late phase. Methods used for release without formal validation, then validated retrospectively under time pressure.
The overall quality control infrastructure supporting these methods — analyst training, instrument qualification, audit trails, OOS handling — matters as much as the methods themselves.
5 Common Questions about Analytical Testing Methods in 2026:
- Is ddPCR required, or is qPCR still acceptable for genome titer?
- Both are acceptable. ddPCR is increasingly preferred for new programs because of its precision and absolute quantification, but qPCR remains valid where well-validated. The agency cares about method performance, not the platform per se.
- What full:empty ratio is acceptable?
- There is no single acceptable threshold. Specifications are set based on the process capability and clinical justification. Many programs target ≥80% full, but lower percentages can be acceptable with appropriate dose specification and toxicology coverage. The key is consistency and characterization.
- Do we need a function-based potency assay for IND?
- Not necessarily. A transduction-based assay can support IND if the relationship to mechanism of action is reasonable. Function-based assays are typically expected by Phase 2/3 if the transduction surrogate doesn’t directly reflect therapeutic mechanism.
- How variable can a cell-based potency assay be?
- Cell-based assays typically show higher variability than chemistry-based assays — 15-30% relative standard deviation is common. Validation should characterize this variability and set specifications accordingly. Excessive variability is sometimes addressable through assay optimization (cell line, reference standard, multiple replicates).
- What’s the most common late-stage CMC issue we should anticipate?
- Method comparability across labs and across reference standard generations. Methods that worked well in a single development lab often show variability when transferred to a CDMO QC lab or a commercial QC site. Build in transfer qualifications early, not as the last activity before PPQ.
Our TLDR:
For an AAV gene therapy program, the analytical strategy is not a downstream concern — it’s the foundation on which every dose, every release decision, and every comparability assessment rests. Vector genome titer methods, capsid characterization, and potency assays should be developed with the rigor and forethought that the long-term program requires, with method validation and reference standard strategy planned years in advance of commercial.
DES Pharma Consulting supports AAV and gene therapy sponsors in scoping, developing, and validating analytical control strategies aligned to current FDA expectations. If your program is preparing for IND, Phase 2 transition, or BLA-enabling validation, this is foundational work that benefits from getting the strategy right early.

Alex has 20+ years of experience in the CMC space, specializing in CDMO/CRO management, analytical development, technology transfer, quality and regulatory compliance for various drug modalities across multiple product stages.
Reach out to Alex on LinkedIn.


