


Pharmaceutical quality assurance continues its evolution toward data-driven decision-making, process analytical technology, and risk-based validation approaches.
Recent regulatory guidance from FDA and EMA provides a clear framework for three technology areas that quality organizations have been developing: 1) artificial intelligence validation and governance, 2) digital reference materials integrated with computer software assurance, 3) and real-time release testing supported by continuous manufacturing principles.
Quality directors and program leaders can implement these capabilities within existing ICH Q10 quality management systems by understanding how current FDA guidances, EMA reflection papers, and ICH Q13 requirements apply to their specific manufacturing contexts and product portfolios.
Understanding Pharmaceutical Compliance: The Foundation
Compliance in pharma refers to adherence to regulatory standards, guidelines, and legal requirements that govern drug development, manufacturing, and distribution. These requirements ensure that pharmaceutical products meet established quality, safety, and efficacy standards before reaching patients.
The regulatory framework encompasses multiple compliance types. Quality compliance addresses adherence to Good Manufacturing Practices (GMP) and quality management system requirements. Regulatory compliance focuses on meeting jurisdiction-specific requirements from bodies like the FDA, EMA, and other national regulatory authorities.
Data compliance has emerged as increasingly critical, encompassing requirements like FDA 21 CFR Part 11 for electronic records and signatures.
What are the Four Phases of Compliance?
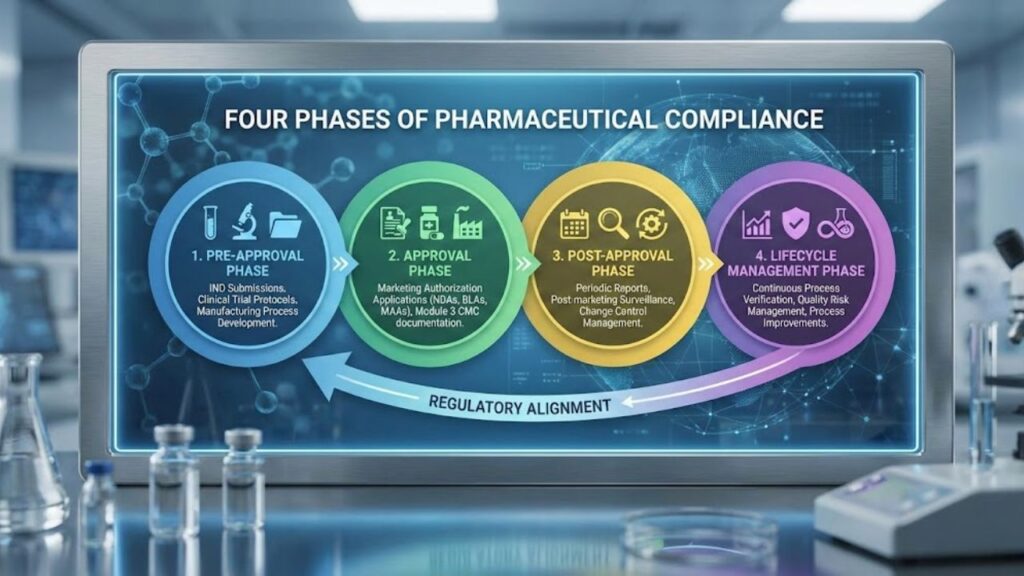
Managing regulatory compliance in pharmaceuticals follows a structured lifecycle with four distinct phases.
- The pre-approval phase encompasses IND submissions, clinical trial protocols, and manufacturing process development.
- The approval phase involves marketing authorization applications (NDAs, BLAs, MAAs) with Module 3 CMC documentation demonstrating process capability.
- The post-approval phase requires ongoing compliance through periodic reports, post-marketing surveillance, and change control management.
- The lifecycle management phase involves continuous process verification, quality risk management, and process improvements while maintaining regulatory alignment.
AI Validation and Governance: The New Regulatory Framework
January 2026 marked an important step toward AI regulation. The FDA and EMA jointly released their Guiding Principles of Good AI Practice in Drug Development. The outlined 10 principles that are intentionally high-level and non-prescriptive. They appropriated aspirational concepts like “human-centric design” and “risk-based approach” without providing concrete validation methodologies or acceptance criteria.
The flexibility in the announcement allows the principles to remain relevant as AI technology evolves, but leaves pharma companies without clear guidance on how to validate AI systems for quality compliance (GMP adherence and batch release decisions), regulatory compliance (IND/BLA submission data), or data compliance (21 CFR Part 11 electronic records and audit trails).
AI validation presents fundamental challenges across all three compliance types (quality, regulatory, and data compliance). Generative AI models, increasingly used for literature review, protocol generation, and regulatory intelligence, can produce different outputs when given identical inputs.
This characteristic is incompatible with traditional validation requirements for reproducibility and deterministic behavior. Large language models exhibit user satisfaction bias, where the AI optimizes responses to please users rather than provide technically accurate information, potentially leading to confirmation bias in safety assessments or efficacy interpretations.
Perhaps most problematically, the “black box” nature of neural networks and transformer architectures makes traditional validation methodology—where you document inputs, processing logic, and expected outputs—nearly impossible to implement with the rigor regulators expect for GxP systems.
These validation challenges explain why the FDA and EMA principles emphasize documentation of data sources, bias mitigation strategies, and ongoing performance monitoring rather than prescribing specific validation protocols.
The principles acknowledge that AI systems require different validation paradigms than deterministic software, focusing on demonstrating fitness-for-purpose through extensive testing across diverse scenarios, continuous monitoring of real-world performance, and human oversight at critical decision points.
Until regulators issue more specific guidance—likely through jurisdiction-specific implementation documents—companies must develop risk-based validation strategies that address stochastic behavior, bias detection, and model transparency within their existing quality management systems.
The FDA’s Considerations for the Use of Artificial Intelligence to Support Regulatory Decision-Making for Drug and Biological Products guidance provides the operational framework for implementing AI within the paradigm of the new regulatory framework. The guidance introduces a seven-step credibility assessment framework that companies must apply when using AI to generate data or information supporting regulatory submissions.
The framework begins with defining the question of interest and establishing the context of use (COU). Risk assessment combines model influence (how much weight the AI output carries) with decision consequence (the severity of potential harm from incorrect predictions).
The credibility plan must be commensurate with this risk level, potentially requiring extensive validation datasets, head-to-head comparisons with established methods, and prospective verification in clinical settings for high-risk applications.
For pharmaceutical quality assurance teams, these frameworks create both obligations and opportunities. Any AI system generating data for regulatory decision-making requires validation following credibility assessments. This includes AI used for analytical method development, process optimization, quality control trending, or stability prediction.
The aforementioned guidances are just a couple examples of the FDA’s and EMA’s explicit recognition that properly validated AI can accelerate development timelines and improve decision quality.
Predetermined Change Control Plans (PCCP): Managing AI Evolution
The August 2025 FDA guidance Marketing Submission Recommendations for a Predetermined Change Control Plan for AI-Enabled Device Software Functions introduces a novel regulatory pathway particularly relevant for AI applications in pharmaceutical manufacturing and quality systems. The PCCP concept offers a blueprint for managing AI systems that learn and evolve post-deployment.
Traditional change control requires regulatory approval for any modification that could significantly affect safety or effectiveness, creating a paradox for AI systems designed to continuously improve.
A PCCP resolves this by establishing, at initial approval, the boundaries of acceptable modifications, the methodology for implementing them, and the validation requirements for verifying they remain safe and effective.
For pharmaceutical applications, this might apply to AI systems controlling continuous manufacturing processes, optimizing bioreactor conditions, or predicting product stability.
The PCCP defines the range of modifications (e.g., retraining with additional batches while maintaining the same algorithm architecture), validation protocols for each modification type, and monitoring systems ensuring the updated model performs as intended.
Digital Reference Materials: The Future of Quality Control
Digital reference materials (dRMs) represent one of pharmaceutical quality’s most transformative innovations, fundamentally changing how companies demonstrate material identity and purity.
Unlike traditional Certificates of Analysis requiring manual transcription into quality systems, dRMs provide structured data in formats like XML, JSON, and AnIML that integrate seamlessly with LIMS, electronic laboratory notebooks, and chromatography data systems.
A dRM for a biologics reference standard might include the complete amino acid sequence, verified spectral data (LC-MS, NMR, IR), validated purity values with method parameters, and bioassay results with acceptance criteria—all cryptographically signed with metadata capturing instrument configuration, analyst identity, and calibration status.
This digitalization delivers multiple benefits. Automated quality checks eliminate transcription mistakes that plague paper-based CoA systems.
Real-time validation occurs at the point of use, with the analytical system automatically verifying that the reference material meets specifications before running samples, rather than discovering discrepancies during batch record review days later.
The FDA’s KASA (Knowledge-Aided Assessment and Structured Application) initiative explicitly supports dRM integration. KASA envisions regulatory submissions where critical quality data exists as structured, searchable datasets rather than static PDF documents.
For BLA submissions, Module 3 (Quality) could include dRM packages that regulatory reviewers query to assess comparability, trending, or specifications alignment. Particularly as the FDA begins to leverage AI further within it’s own review processes.
For chromatographic analyses, dRM implementation faces unique challenges due to method-specific variability in column chemistry, mobile phase composition, gradient programs, and detector settings.
Addressing this requires flexible data models accommodating method-specific parameters, robust metadata standards, and inter-laboratory calibration protocols ensuring dRM comparability across different chromatographic systems.
Computer Software Assurance: Modernizing Validation
The September 2025 FDA guidance Computer Software Assurance for Production and Quality System Software represents a fundamental shift in how pharmaceutical manufacturers validate software systems.
This guidance replaces documentation-heavy Computer System Validation with a risk-based, critical-thinking framework emphasizing fitness-for-purpose over prescriptive testing protocols.
The CSA guidance applies to software used as part of pharmaceutical device production or quality systems—including LIMS, manufacturing execution systems, electronic quality management systems, and document management platforms.
It explicitly covers cloud-based solutions (IaaS, PaaS, SaaS), ending ambiguity about whether cloud-based quality systems require the same validation rigor as on-premise installations.
The framework’s risk-based approach classifies software features as either high process risk (where failure could foreseeably compromise patient safety) or not high process risk. Validation activities scale to risk level. High-risk functions require scripted testing with documented test cases.
Lower-risk functions might undergo unscripted exploratory testing. Continuous monitoring through system logs and performance metrics can supplement or replace traditional testing for certain low-risk functions.
The guidance explicitly endorses leveraging vendor evidence rather than duplicating validation activities. If a SaaS vendor provides ISO 27001 certification, SOC 2 Type II reports, validated software development lifecycle documentation, and customer acceptance testing evidence, pharmaceutical companies can incorporate these into validation packages rather than re-testing vendor-validated functionality.
For pharmaceutical quality assurance teams, CSA implementation requires both technical and cultural shifts. The technical shift involves documenting intended use for each software system and evaluating which features carry high process risk. The cultural shift requires quality and IT teams to apply critical thinking rather than following prescriptive checklists. Validation doesn’t mean perfect—it means fit-for-intended-use with appropriate controls for identified risks.
Real-Time Release Testing: From Lab to Manufacturing Floor
Real-time release testing represents a paradigm shift from traditional release strategies where products await laboratory testing results. RTRT uses process data and in-process measurements to demonstrate finished product quality at the time of manufacture, enabling immediate release without retained sample testing.
ICH Q13 Continuous Manufacturing of Drug Substances and Drug Products, finalized in November 2022 and implemented by FDA in March 2023, provides the regulatory framework supporting RTRT for continuously manufactured products. Continuous manufacturing involves continuous feeding of input materials, transformation of in-process materials, and concomitant removal of output materials—fundamentally different from batch manufacturing’s discrete, sequential operations.
The guidance emphasizes that CM requires enhanced process understanding. Companies must demonstrate process residence time distribution, understand how transient events (start-up, shutdown, flow rate changes) affect product quality, establish control strategies managing variability in real-time, and implement robust diversion strategies ensuring out-of-specification material never reaches finished product.
RTRT implementation requires validating that process measurements reliably predict finished product quality.
This might involve demonstrating correlation between in-line NIR spectra and tablet content uniformity, showing that continuous TOC monitoring predicts pharmaceutical water purity, or validating that bioreactor metabolite profiles predict final product quality attributes.
ASTM E2656 Standard Practice for Real-time Release Testing of Pharmaceutical Water for the Total Organic Carbon Attribute provides a practical template for RTRT implementation. The standard establishes risk-based approaches scaling validation effort to the consequence of failing TOC specifications, methodologies for qualifying online TOC instrumentation, sampling frequency determination, and documentation requirements supporting regulatory inspection.
Key to successful water RTRT is demonstrating that online TOC data provides equivalent or superior quality assurance compared to laboratory grab samples.
This requires addressing instrument delay time, establishing appropriate alarm setpoints triggering diversion, maintaining the online analyzer in a validated state, and integrating TOC data into batch records through automated data acquisition.
Modern microbial methods extend RTRT principles to microbiological quality control, addressing one of pharmaceutical manufacturing’s most significant time bottlenecks.
Traditional microbial enumeration requires 3-7 days for colony formation. Modern methods including flow cytometry, ATP bioluminescence, PCR-based methods, and MALDI-TOF mass spectrometry reduce time-to-result from days to hours or minutes.
The M3 Collaboration documented how these methods support contamination control strategies required by EU GMP Annex 1.
Modern methods enable real-time contamination detection in pharmaceutical water systems, rapid bioburden assessment of raw materials, continuous environmental monitoring in aseptic processing areas, and accelerated investigation of contamination events.
Integration: Building a Digital Quality Ecosystem
The true power of these emerging technologies emerges through integration rather than isolated implementation. AI credibility assessment frameworks validate algorithms that process data from digital reference materials.
Computer software assurance principles govern the quality systems managing AI models, dRM databases, and RTRT infrastructure. Real-time release testing generates the high-frequency data streams that train AI models and validate digital twin predictions.
This integration requires pharmaceutical companies to think systemically about quality infrastructure.
Successful integration requires defining data standards enabling interoperability between analytical instruments, quality systems, and manufacturing platforms; establishing governance frameworks with clear ownership for AI models, digital systems, and data quality; investing in workforce capability development; and maintaining regulatory alignment through proactive engagement with FDA, EMA, and other authorities.
The FDA’s KASA initiative and eCTD 4.0 structured submission formats anticipate this integrated future. Companies building integrated digital quality ecosystems now position themselves to leverage KASA’s benefits while competitors struggle retrofitting legacy systems into structured formats.
5 Strategic Recommendations for 2026
Quality leaders should prioritize several strategic actions in 2026.

- First, assess AI readiness by conducting inventories of current AI applications, evaluating existing data infrastructure and governance, and establishing cross-functional AI oversight committees. Many companies unknowingly use AI today—statistical process control software, predictive maintenance algorithms, or automated image analysis tools often embed machine learning.
- Second, establish dRM roadmaps by identifying analytical methods and reference materials benefiting most from digitalization, engaging with LIMS and CDS vendors about dRM integration capabilities, and participating in industry working groups (PDA, USP, BioPhorum) shaping dRM standards.
- Third, modernize software validation by reviewing existing CSV practices identifying disproportionate effort on low-risk systems, training quality and IT teams on CSA principles, and piloting CSA approaches for new system implementations before attempting legacy system remediation.
- Fourth, develop RTRT strategies by evaluating where RTRT delivers greatest value, investing in PAT infrastructure and process understanding enabling real-time quality assessment, and exploring modern microbial methods for applications where traditional testing creates bottlenecks.
- Finally, engage regulators early through pre-IND meetings discussing AI validation strategies, scientific advice procedures seeking feedback on digital twin approaches, and Q-Submission programs obtaining input on PCCP proposals for evolving AI systems.
The Compliance Framework for Digital Transformation
Successful pharmaceutical compliance in 2026 requires understanding that technology adoption must align with regulatory fundamentals.
The pharma compliance framework hasn’t changed—products must still be safe, effective, and consistently manufactured to approved specifications. What’s changing is how companies demonstrate compliance through structured data, validated AI models, and real-time quality assurance.
This compliance certification path begins with establishing robust quality management systems following ICH Q10 principles, integrated with new technological capabilities.
Companies must maintain comprehensive documentation—now including AI validation reports, dRM specifications, and RTRT protocols—accessible for regulatory inspections. Managing regulatory compliance in the digital era means demonstrating data integrity throughout the quality lifecycle.
The importance of compliance in the pharmaceutical industry intensifies as quality systems grow more complex. Traditional batch-based quality control created natural review points where human judgment verified product quality.
Digital quality systems with AI-driven decisions and automated release create different vulnerabilities—algorithm errors, data integrity failures, or cyber intrusions that might not be immediately apparent.
Regulatory guidelines are evolving to address these challenges. The list of regulatory guidelines for pharmaceuticals now explicitly includes AI-specific guidance, digital quality guidance, continuous manufacturing guidance, and data integrity guidance.
Companies must maintain awareness of these evolving requirements, updating quality systems and training programs as guidance finalizes.
The transition to digital quality systems shouldn’t be perceived as a compliance burden but rather as an opportunity to enhance pharmaceutical quality while improving operational efficiency. AI-validated analytical methods reduce false positive investigations.
Digital reference materials eliminate transcription errors causing manufacturing delays. Real-time release testing accelerates product availability while reducing inventory carrying costs. Computer software assurance focuses validation effort where it matters most.
Our Final Thoughts: Positioning for Success in the Digital Quality Era
Pharmaceutical quality assurance in 2026 stands at an inflection point. The regulatory frameworks governing AI validation, digital quality systems, and real-time release testing are now established. Companies that engage with these frameworks strategically—investing in data infrastructure, building cross-functional capabilities, modernizing validation practices, and maintaining regulatory dialogue—position themselves as digital quality leaders.
The question for quality leaders isn’t whether to embrace AI, digital twins, and real-time release testing, but rather how to implement these technologies in ways that maintain regulatory compliance while delivering business value.
The regulatory frameworks exist. The technological capabilities are proven. The implementation roadmaps are clear. What’s required now is leadership commitment, strategic investment, and organizational capability development.
For biopharmaceutical companies developing advanced therapies—AAV gene therapies, monoclonal antibodies, CAR-T cell therapies—these digital quality capabilities aren’t nice-to-have enhancements but competitive necessities.
The analytical complexity, manufacturing challenges, and regulatory expectations for these modalities demand sophisticated quality systems leveraging AI for process optimization, digital twins for process understanding, and real-time monitoring for contamination control.
As your organization navigates this transition, building internal capabilities while maintaining regulatory compliance will determine success in the evolving pharmaceutical quality landscape.
For expert level pharmaceutical consulting, get in touch with us at DES Pharma for a free initial consultation.

Senior Consultant, Process Development & CMC



