


Biotech startups operating under FDA-regulated conditions face a deceptively simple question: when we adopt a cloud-based quality management system, what do we actually have to do to be 21 CFR Part 11 and ALCOA+ compliant?
The answer is more nuanced than vendors typically present. A SaaS QMS being “Part 11 compliant” is necessary but not sufficient — the regulation places obligations on you, the regulated entity, that no vendor can satisfy on your behalf. This guide walks through what’s actually required, what’s commonly misunderstood, and how to scope a validation program that holds up under FDA inspection.
Why this matters more for startups than people think
Established pharma companies have decades of validated systems, internal QA infrastructure, and dedicated computer system validation (CSV) staff. Startups don’t. The temptation is to defer “real” QMS validation until later — pick a cloud QMS, sign the order form, start using it, and worry about formal validation when an audit looms.
This is a costly mistake for three reasons:
- Pre-IND and IND-enabling activities generate regulated records. If your QMS holds GLP toxicology study documentation, GMP manufacturing records, or any data supporting an IND submission, Part 11 applies the moment those records are created — not when you submit.
- Validation gaps compound. Records created in an unvalidated system don’t become validated retroactively. The longer you operate without proper validation, the larger the remediation burden becomes.
- FDA inspections increasingly focus on data integrity. ALCOA+ deficiencies are among the most common findings in FDA 483s and warning letters over the last five years, and investigators specifically probe how cloud systems are validated and controlled.
What 21 CFR Part 11 actually requires
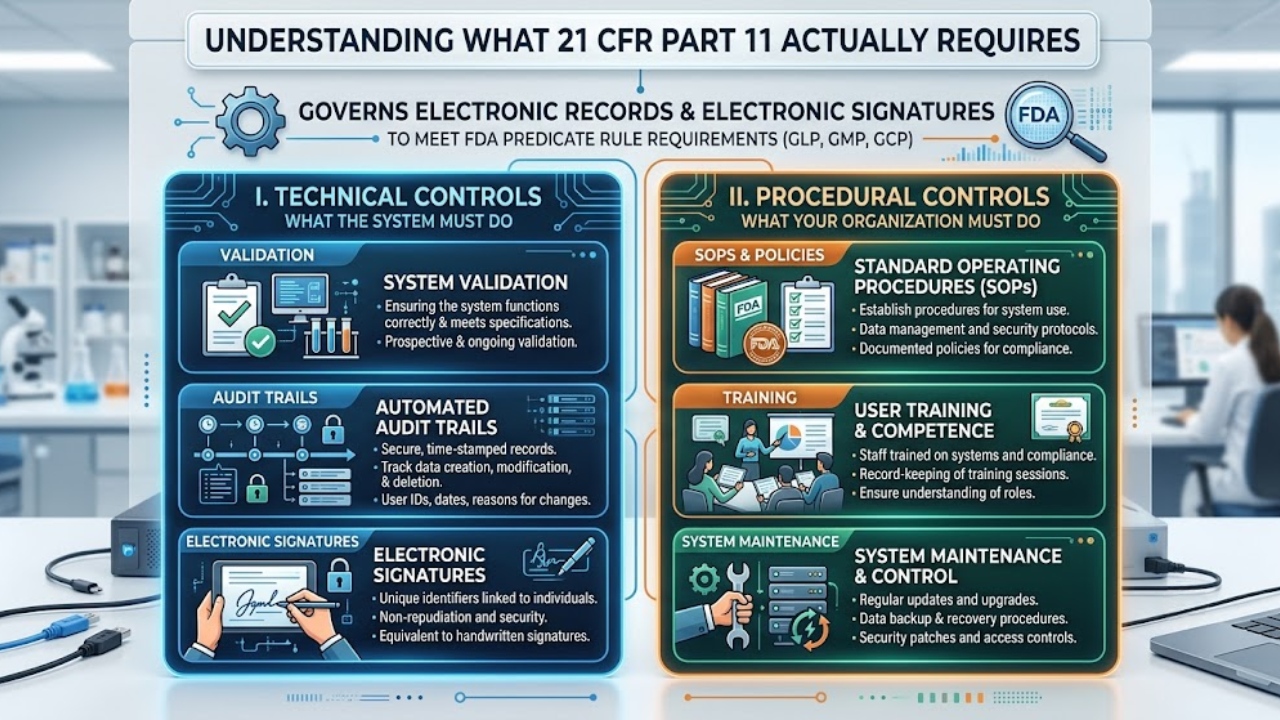
21 CFR Part 11 governs electronic records and electronic signatures used to meet FDA predicate rule requirements (GLP, GMP, GCP). The regulation has two functional halves: technical controls (what the system must do) and procedural controls (what your organization must do).
Technical controls Part 11 requires
- Validation of systems to ensure accuracy, reliability, and consistent intended performance
- Ability to generate accurate and complete copies of records (human-readable and electronic)
- Protection of records throughout the retention period
- Limited access to authorized individuals
- Secure, computer-generated, time-stamped audit trails
- Operational system checks to enforce sequencing of steps
- Authority checks to ensure only authorized individuals can use the system
- Device checks where appropriate
- Persons who develop, maintain, or use the system must have appropriate education, training, and experience
- Written policies that hold individuals accountable for actions initiated under their electronic signatures
- Appropriate controls over systems documentation
Electronic signature requirements
- Each signature must be unique and not reused or reassigned
- Identity verification before signature credentials are issued
- Two-component authentication for non-biometric signatures (typically user ID + password)
- Signed records must include printed name, date and time, and meaning of the signature
- Signatures must be linked to records such that they cannot be excised, copied, or transferred
- Submission of a letter of certification to FDA stating that electronic signatures are intended to be legally binding equivalents of handwritten signatures
ALCOA+ and what it adds
ALCOA+ is the data integrity framework FDA and international regulators apply when evaluating whether records — paper or electronic — are trustworthy. The acronym expands on the original ALCOA principles:
| Principle | What it means in a cloud QMS context |
|---|---|
| Attributable | Every action is tied to a uniquely identified user via authenticated login |
| Legible | Records remain readable in human-readable form throughout retention |
| Contemporaneous | Actions are timestamped at the moment they occur, not after the fact |
| Original | The first record (or certified true copy) is preserved with full audit trail |
| Accurate | Records reflect what actually happened, with no unauthorized edits |
| + Complete | All data including metadata and repeat analyses are retained |
| + Consistent | Records are in expected chronological sequence with no gaps |
| + Enduring | Records survive for the full required retention period in usable form |
| + Available | Records are retrievable on demand throughout retention |
ALCOA+ is not codified in a single regulation — it’s a framework regulators apply when inspecting how you meet Part 11, EU Annex 11, and predicate GMP/GLP rules. A robust quality assurance program bakes ALCOA+ into SOPs, training, and system design rather than treating it as an afterthought.
The shared responsibility model nobody explains clearly
Here’s the part vendors gloss over: when you adopt a cloud QMS, validation and compliance responsibilities are shared between the vendor and you. The vendor handles infrastructure and the platform itself; you handle configuration, business processes, and use.
| Area | Vendor responsibility | Your responsibility |
|---|---|---|
| Infrastructure | Data center physical security, network security, hypervisor patching, backup infrastructure | Vendor qualification audit, ongoing oversight, SLA monitoring |
| Platform | Platform development under SDLC, change control, internal validation testing, security patches | Review of vendor’s validation evidence, qualification of platform for your use |
| Configuration | Documentation of configurable options | Configuration specification, configuration testing, configuration change control |
| Business processes | None | SOPs, workflow design, training, user access governance, signature manifestations |
| Data | Encryption at rest and in transit, backup execution | Data ownership, retention policy, periodic restore testing, archival on contract termination |
FDA does not accept “the vendor said they’re Part 11 compliant” as a defense. You are the regulated entity. You must demonstrate that the system, as configured and used in your environment, meets the regulation.
Vendor qualification: what to actually look for
Before you select a cloud QMS, conduct a vendor qualification. At minimum, the vendor should provide:
- SOC 2 Type II report — covering at least Security, Availability, and Confidentiality trust services criteria, with a recent audit period
- ISO 27001 certification for information security management (strongly preferred)
- Validation package / qualification documentation — IQ/OQ evidence the vendor performs on each release, including test scripts and traceability matrices
- SDLC documentation — how the vendor controls software development, including change management, code review, and release procedures
- Audit trail specifications — what’s captured, where it’s stored, how it’s protected from modification, and how it can be exported
- Subprocessor list — most cloud QMS vendors use AWS, Azure, or GCP as subprocessors; you need to understand the chain
- Data residency and retention policies — where data is stored, how it’s deleted, what happens on contract termination
- Incident response and breach notification commitments in writing
- Quality agreement — a separately executed agreement that defines GxP roles, responsibilities, and notification obligations (this is non-negotiable for serious work)
Do not accept marketing claims of “Part 11 compliant” without evidence. Ask for the validation documentation. Ask to see audit trail samples. Ask what happens to your data if they go out of business. Reputable vendors will provide these readily; the ones that won’t are telling you something.
Scoping your validation: a risk-based approach
FDA’s general principles of software validation and the GAMP 5 framework both endorse a risk-based approach. You don’t need to validate every feature equally — you validate based on the regulatory risk of the records and processes the system supports.
A pragmatic scoping approach for a biotech startup:
- Identify GxP scope. Which modules will hold GxP records? Document control, training records, deviations, CAPAs, change control, audit management, supplier management are the typical first wave. Out-of-scope modules (e.g., HR document templates that are not regulated records) can be excluded from validation.
- Categorize functions by risk. Workflows that create or modify regulated records, manage electronic signatures, or control access are high-risk. Reporting and read-only views are lower risk.
- Plan testing depth by risk. High-risk functions get scripted PQ (performance qualification) testing with documented evidence. Lower-risk functions can be tested at the OQ level using vendor-supplied test scripts.
- Document configuration. Every configurable option that affects GxP behavior — user roles, workflow rules, signature requirements, audit trail settings — must be specified and tested as configured.
The validation deliverables you actually need
For a cloud QMS at a biotech startup, a defensible validation package typically includes:
- Validation Plan — scope, approach, risk assessment, roles, deliverables, acceptance criteria
- User Requirements Specification (URS) — what the system must do for your processes
- Functional Risk Assessment — risks identified and categorized, with mitigation tied to validation activities
- Configuration Specification — every configurable parameter documented as configured
- IQ (Installation Qualification) — typically leveraged from vendor documentation for SaaS
- OQ (Operational Qualification) — verification that platform features work as specified (often leveraged from vendor)
- PQ (Performance Qualification) — verification that the system, as configured, supports your specific workflows correctly
- Traceability Matrix — linking URS items to risk assessment to test scripts to test results
- Validation Summary Report — conclusions, residual risks, and statement of validated status
- Ongoing controls — periodic review SOP, change control procedure, vendor oversight procedure, user access review procedure
Procedural controls: the part that fails inspections
Technical Part 11 compliance is generally well-handled by reputable vendors. Procedural controls are where startups fail. Specifically, you need written SOPs and evidence of execution for:
- User access management. How are users provisioned, modified, and deprovisioned? How quickly are departing employees removed? Quarterly user access reviews are the standard expectation.
- Electronic signature policies. Training records, signed acknowledgments that users understand their signatures are legally binding, the letter of certification submitted to FDA.
- Change control. Both for vendor-driven platform changes (releases) and for your own configuration changes.
- Periodic review. Annual (at minimum) review of system performance, audit trail spot-checks, user access, and continued fitness for purpose.
- Training. Documented training on the system, on Part 11, and on ALCOA+ for all users.
- Backup and disaster recovery testing. Even if the vendor performs backups, you should periodically verify you can retrieve records.
- Incident management. Procedures for handling deviations, breaches, and integrity events involving the QMS.
Common deficiencies and how to avoid them
The most frequent findings in cloud QMS validation audits and FDA inspections:
- Inadequate vendor qualification. Selection based on price and demo without formal qualification documentation.
- “Validation” that is just vendor’s documentation in a binder. No customer-specific PQ, no configuration testing.
- Audit trail not reviewed. Audit trails exist but no one ever looks at them. Periodic audit trail review should be a documented activity.
- Stale user access. Former employees still active in the system, shared accounts, generic admin accounts in use.
- Configuration drift. System configured one way at validation, evolved over time without controlled change management.
- No periodic review. System validated at go-live, never re-evaluated. Three years later, no one can attest to current validated state.
- Inadequate signature training. Users signing electronically without documented training on signature meaning and legal weight.
Building these controls is unglamorous work, but it’s the difference between a clean audit and a Form 483. A well-structured pharmaceutical quality framework integrates QMS validation into the broader quality system rather than treating it as a standalone IT project.
Timeline and cost for a biotech startup
For a Series A/B biotech adopting a cloud QMS for the first time, expect:
- Vendor selection and qualification: 6-10 weeks
- Implementation and configuration: 8-16 weeks depending on scope
- Validation execution: 6-12 weeks running in parallel with later implementation
- Total elapsed time: 4-7 months from kickoff to validated go-live
- Cost: $40K-$150K for validation services (external) plus internal effort, on top of vendor licensing
Doing this right at Series A costs a fraction of remediating it at Series C when due diligence or a pre-approval inspection forces the issue.
Frequently asked questions
- Is “21 CFR Part 11 compliant” software actually compliant out of the box?
- No. Software can be Part 11-capable, but compliance depends on how it’s configured, validated, used, and governed by procedures in your environment. The vendor handles the platform; you handle the rest.
- Do we need a letter of certification to FDA before using electronic signatures?
- Yes. 21 CFR 11.100(c) requires submission of a certification to FDA stating that electronic signatures in your systems are intended to be the legally binding equivalent of handwritten signatures. This is a one-time letter from your organization to FDA, not per-system.
- How does GAMP 5 categorization apply to a cloud QMS?
- A configured cloud QMS is typically GAMP Category 4 (configured product) — meaning you accept the vendor’s validation of the core platform but must validate your specific configuration. Highly customized implementations may approach Category 5, requiring more extensive validation.
- What’s the relationship between Part 11 and EU Annex 11?
- EU Annex 11 covers similar ground for the European regulatory context — computerized systems used in GMP environments. The two are largely compatible; a system designed for Part 11 with appropriate procedural controls generally satisfies Annex 11, though Annex 11 places additional emphasis on supplier risk assessment and IT infrastructure qualification.
- Do we need to validate Microsoft 365 or Google Workspace if we use it for GxP documents?
- If you use general-purpose tools to create, store, or sign GxP records, those tools fall in scope of Part 11. This is one reason a dedicated QMS is strongly preferred over ad-hoc use of office productivity tools — validating M365 for GxP use is substantially more complex than validating a purpose-built QMS.
Closing thoughts
21 CFR Part 11 and ALCOA+ compliance for a cloud QMS is not optional, not deferrable, and not something a vendor can deliver to you in a box. It requires deliberate scoping, a real vendor qualification, a customer-specific validation, and ongoing procedural discipline. For biotech startups, the right time to do this work is when you first adopt the system — not when an inspector asks.
DES Pharma Consulting helps emerging biotechs scope and execute defensible computer system validation programs for cloud QMS and other GxP systems. If you’re approaching IND-enabling work, this is foundational infrastructure that benefits from getting right the first time.

Alex has 20+ years of experience in the CMC space, specializing in CDMO/CRO management, analytical development, technology transfer, quality and regulatory compliance for various drug modalities across multiple product stages.
Reach out to Alex on LinkedIn.



